On average, the process of bringing a drug from initial laboratory research to FDA approval requires 10 to 15 years and costs over $2 billion, a timeline that has remained static for decades despite significant advances in compute. The discovery phase, which involves target identification, validation, and the design of molecules with appropriate affinity, selectivity, and drug-like properties, typically takes up the first four to six years of development and is, by almost any measure, the most intellectually demanding stage in all of pharmaceutical R&D. PitchBook has argued that AI could materially improve overall clinical success rates from about 7.9% to 17.7%, with the largest gains in early clinical phases. AI-native biotechs have so far achieved approximately 80 to 90% Phase I success rates compared to the industry average of 40 to 65%, and 40% in Phase II versus the historical average of 29%, although the dataset remains small at roughly 10 completed trials. This piece maps the landscape of AI-driven drug discovery: what these tools actually do, how they work at a level useful for decision-making, and where they fit into the broader role of AI integration in the biopharma industry.
AI Scientists and Analysts
The term "AI scientist" gets thrown around loosely; ultimately, companies building in this space aim to answer a simple question: can an AI system autonomously conduct the intellectual work of scientific research: reading literature, forming hypotheses, designing experiments, interpreting results, and iterating? FutureHouse is one of the clearest examples of the AI scientist thesis in biotech, and its trajectory over the past year illustrates just how quickly this space is evolving. In September 2024, FutureHouse launched PaperQA2, a literature-retrieval and synthesis agentic RAG system built to search papers, follow citations, extract evidence, and iteratively refine answers. Core to PaperQA2's step-change improvement over previous models was its agentic nature: after a first round of search and evaluation, the model autonomously decides whether to run additional searches, reformulate the query, or revise the structure of its answer.
PaperQA was rebranded as Crow in May 2025, and became just one cog in an open platform of specialized agents for distinct research tasks: Crow, Falcon, and Owl for literature search and synthesis, Phoenix for chemistry planning, and Finch for complex data analysis. FutureHouse then integrated a subset of those capabilities into a multi-agent workflow called Robin in its first published therapeutic case study. Tasked with finding novel therapeutics for dry age-related macular degeneration (AMD), Crow's rigorous literature review led to the hypothesis that improving retinal pigment epithelium phagocytosis could help dry age-related macular degeneration. Robin first surfaced Y-27632 as a promising ROCK inhibitor, then proposed an RNA-seq follow-up. Finch interpreted the sequencing data to connect the effect to ABCA1 upregulation, and Robin then proposed a second round of candidates, which led to ripasudil, a Japanese glaucoma drug, as a top hit.
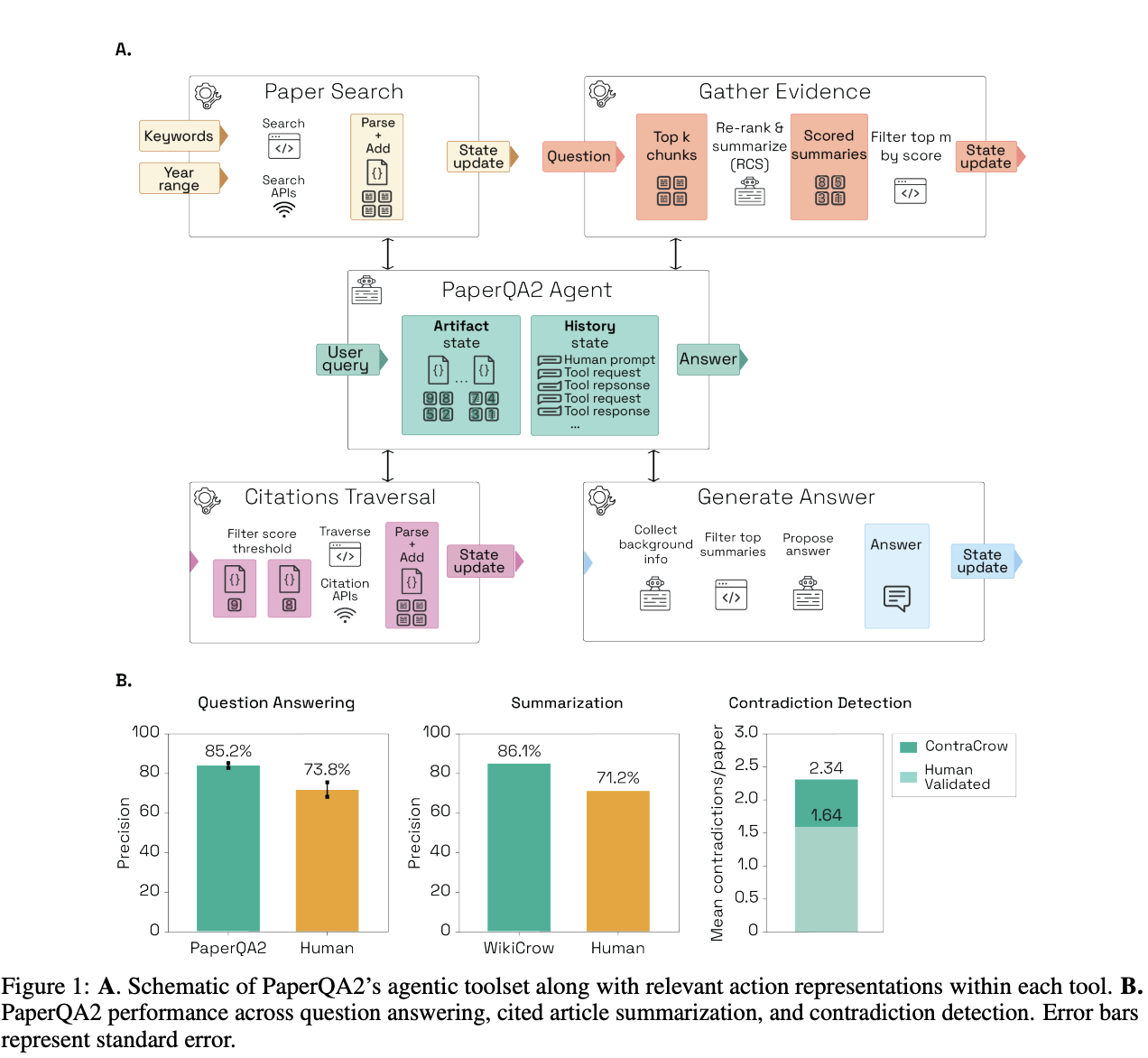
The FutureHouse case is important for two reasons beyond the specific discovery. First, Robin represents one version of the lab-in-the-loop model I expect to dominate the future of AI-augmented research: while agents own the intellectual steps (hypothesis generation, experiment design, data analysis), human researchers use their intuition and domain expertise to validate and question the AI output and execute physical experiments. Second, the sheer speed of iteration is astounding: the entire process from conceptualizing Robin to paper submission was completed in roughly 2.5 months by a small team.
AI scientists have also played an outsized role in target ID and validation, automating the first critical decision in drug discovery: which biological target should we go after? One of the most instructive case studies for the role of AI in target ID is Insilico Medicine's TNIK fibrosis program. The company's proprietary platform, PandaOmics, took in multiomics datasets from IPF patient tissue, then contextualized omics-derived hypotheses through biological network analysis and text evidence from clinical trials, publications, and grant applications, controlling for "node degree bias," preventing the model from over-weighting particular pathways and genes simply because they appear in more datasets. PandaOmics' preprocessing layer includes dataset selection, case/control grouping, dimensionality reduction (PCA/tSNE/UMAP), batch correction/QC, and even mapping methylation to genes. Notably, scientists can tune the model to prioritize "high-confidence targets," those validated in both omics data and external literature, as well as "novel targets," those unearthed exclusively by analysis of omics data.
Insilico's 2024 validation paper in Nature Biotechnology described target identification as a ranked dashboard where each column is a separate disease-specific model. On the omics side, PandaOmics runs multiple parallel workflows: direct expression/proteomics/methylation analysis; identification of neighbors on protein-interaction graphs; causal inference based on transcription-factor/regulatory-network enrichment; "HeroWalk," a stochastic graph traversal model that learns embeddings (vectors) for genes and diseases; matrix factorization, which starts with a sparse gene-disease association matrix and factorizes it into lower-dimensional latent factors; mutation burden; overexpression and knockout concordance; and a "relevance score" tied to known target/clinical-trial evidence. The current PandaOmics docs describe the addition of text scores such as attention, trend, attention spike, and evidence, plus financial, KOL, and LLM-derived scores for confidence, druggability, mechanism clarity, and commercial tractability. The final ranking is therefore produced by re-weighting and filtering these scores for the context of a scenario-specific decision.
TNIK emerged as the number-one candidate after Insilico applied filters for disease mechanisms, protein class, and small-molecule druggability. From there, the company's Chemistry42 platform generated the lead compound, INS018_055, now called rentosertib (note the convergence of the AI scientist and molecule designer under one company umbrella: an increasingly common theme). The entire process from target discovery to preclinical candidate nomination took 18 months, compared to the typical four to six years; Rentosertib is now in Phase 2a clinical trials.
Benchling, the dominant R&D cloud platform used by over 1,300 biotech and pharma companies including Moderna, Sanofi, and Eli Lilly, has released a deep research agent that operates over Benchling data with the context of the Benchling data model, enabling scientists to ask complex cross-dataset questions that previously took weeks or months and get answers in hours. CEO Sajith Wickramasekara gives a specific example of how Benchling's agentic infrastructure saved one customer nearly eight months of work: a team preparing to run mouse studies across 20 models used the deep research agent to discover that a significant subset of those experiments had already been run years earlier by scientists at a company that had since been acquired. Benchling is also integrating open-source and proprietary models (including Boltz, Chai, and AlphaFold) directly into scientific workflows, pre-configured so that wet lab scientists without computational skills can run simulations and have the results link automatically to their existing data in Benchling. The longer-term aspiration is to move from passive simulation toward active recommendation: suggesting the next best experiment based on a scientist's historical work and the public literature. Its Experiment Optimization feature trains a "tournament" of ML models on a scientist's historical data to identify which input parameters most influence outcomes, then uses Bayesian optimization to recommend specific new experimental conditions most likely to improve the target variable in the next iteration. The company is now tightening the integration between Experiment Optimization and study planning, and through a partnership with Stanford's Generative Expert Labs (GXL), is building domain-specific expert agents that reason across datasets and recommend model runs natively inside Benchling.
Molecule Design
The evolving integration of AI in the drug discovery process can be split into two distinct eras:
Era 1 (2020-2023): Structure prediction. AlphaFold2 solved a 50-year-old problem by accurately predicting what proteins look like in 3D from their amino acid sequences. The 2024 Nobel Prize in Chemistry went jointly to David Baker (for computational protein design) and Demis Hassabis/John Jumper (for AlphaFold). This was the solution to the age-old "protein folding problem" that was absolutely transformative for understanding biology. But with a key caveat: solving for the physical representation of a sequence didn't design new drugs.
Era 2 (2024-present): Generative design. A new generation of models can now propose entirely new molecules from proteins to antibodies to small-molecule drugs designed to bind specific targets with high affinity. Generative architectures are far more complex, combining diffusion models, transformers, and graph neural networks (GNNs) into sequential foundation models capable of working across proteins, small molecules, DNA, RNA, and their multifaceted interactions. While dozens of companies are competing in Era 2, three stand out.
Isomorphic Labs: The Full-Stack Pharma Thesis
Isomorphic Labs, Alphabet's autonomous drug discovery subsidiary led by Nobel laureate Demis Hassabis, was spun out of DeepMind in 2021, and has since grown to over 200 employees with partnerships valued at nearly $3 billion. The company was built on the AlphaFold lineage, each generation of which expanded the boundary of what AI could do for biology. AlphaFold2 (2020) predicted what proteins look like; AlphaFold3 (May 2024) predicted how they interact with other molecules; AlphaProteo (September 2024) generated new binding proteins (achieving 3-300x better binding affinities than existing methods across 7 targets, including the first-ever AI-designed binder for VEGF-A). On February 10th, 2026, Hassabis's team published IsoDDE (Isomorphic Drug Design Engine), a unified computational system that moves decisively beyond AlphaFold3 into end-to-end drug design. Isomorphic claims that IsoDDE "more than doubles" AlphaFold 3's accuracy on the "Runs N' Poses" benchmark (Skrinjar et al. 2025), created to determine a model's ability to generalize past its training data to novel pockets and ligands. On antibody-antigen structure prediction, one of the hardest challenges in computational biology because of the flexible, hypervariable CDR-H3 loop (the primary mediator of binding specificity on an antibody heavy chain), IsoDDE reportedly outperforms AlphaFold3 by 2.3x and Boltz-2 by 19.8x (notably, Boltz is a model optimized for small-molecule affinity rather than antibody structure, but the comparison is nonetheless impressive).
Importantly, while companies like Chai Discovery and Boltz position themselves as AI platform companies, Isomorphic's thesis is fundamentally different from its platform counterparts, positioning itself as a bona fide biopharma company advancing its own assets in-house. AlphaFold's structure prediction is only one piece of the Isomorphic equation, as the company layers proprietary chemistry models, potency prediction, and ADMET optimization on top. Its pipeline is currently focused on small molecules, with CEO Hassabis indicating that the company expects its first AI-designed drugs to enter human trials by the end of the year. Isomorphic raised $600 million in its first external funding round (March 2025, led by Thrive Capital), supplementing nearly $3 billion in combined deal potential from partnerships with Eli Lilly ($45M upfront, up to $1.7B milestones) and Novartis ($37.5M upfront, up to $1.2B milestones).
Boltz: The Open-Source Affinity Predictor
Before Boltz-2, if your discovery team wanted to computationally predict binding affinity (how tightly a candidate molecule binds to a target) they had two options. They could use expensive, slow physics-based simulations called free-energy perturbation (FEP) that take hours to days per compound and require specialized computational chemistry expertise, or they could use faster AI models, which, for what they improved upon FEP in speed, lacked in accuracy, too imprecise to rely upon. Boltz-2, developed by MIT Jameel Clinic alongside Recursion, was the first deep learning model to approach FEP-level accuracy in binding affinity prediction while running roughly 1,000 times faster, approximately 18 to 20 seconds per prediction on a single GPU. Architecturally, Boltz-2 is composed of four modules: the ubiquitous Pairformer for molecular representation, a diffusion-based denoising module, a confidence module, and an affinity module. The affinity module is trained on approximately 5 million binding affinity assay measurements curated from ChEMBL, BindingDB, PDBbind, and MF-PCBA, yielding roughly 750,000 high-quality protein-ligand pairs after filtering. Boltz-2's training data is also multimodal, incorporating PDB structures, NMR data, and publicly available molecular dynamics simulations. Unlike Boltz-1, Boltz-2 also includes a layer called "Boltz-steering" that uses physics-based corrections at inference time to avoid proposing molecules that would physically crash into each other (steric clashes), a common failure mode for pure AI approaches.
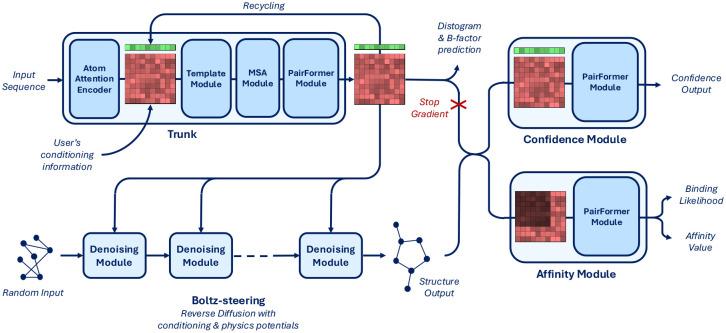
Importantly, another critical differentiator is Boltz-2's licensing: currently, the model is fully open-source under an MIT license, with model weights, training code, and the complete pipeline available for both academic and commercial use. As co-creator Gabriele Corso put it to R&D World magazine, "99.9% of drug developers and biologists are outside of companies like Isomorphic. Part of the reason we are releasing this fully open source is because we want all of these biologists to have access to it."
Chai Discovery: Zero-Shot Antibody Design
Chai-2 is a multimodal generative model for de novo antibody and protein binder design that creates entirely new sequences and structures, not just predicting existing ones. Founded in 2024 by Joshua Meier (who co-led ESM-1 development at Meta FAIR and served as Chief AI Officer at Absci), Chai Discovery went from idea to $1.3 billion valuation by December 2025, barely 18 months after incorporation.
Chai-1, founded in September 2024, was the company's breakout model, building on the core architecture of AlphaFold with a few significant twists, most notably including residue-level embeddings from a 3-billion-parameter protein language model and a modular conditioning system that accepts experimental restraints (pocket conditioning, contact constraints, docking constraints). In simpler terms, this means that for each residue in an input protein sequence, Chai could generate high-dimensional vectors (embeddings) of each amino acid position, using spatial context to encode whether that residue sits in a likely helix or loop, whether nearby residues suggest a binding motif, or whether it engages in any long-range within-protein interactions. Additionally, unlike early AlphaFold models, Chai could be "guided" by external data rather than forced to predict blindly. In many real experimental settings, scientists already know part of the answer: cross-linking data may suggest close contact of two residues in an unknown structure, or prior groups have suggested evidence of a binding pocket. Chai-1 introduced conditioning channels for experimental data, allowing it to update its prediction and shrink its search space.
Chai-2, launched in June of 2025, was an inflection point for the company, as it moved from prediction to generative design. Instead of starting with antibody libraries or known binders, the model takes as input the antigen structure and the specific epitope region that should be targeted, then jointly generates antibody sequence and structure, particularly the complementarity-determining regions (CDRs) that form the binding interface. Across 52 novel targets with unknown binders, Chai-2 achieved a 16% overall hit rate for de novo antibodies, ~20% for nanobodies, and a striking 68% hit rate for miniprotein binders with picomolar affinities. The system even designed functional GPCR agonists, historically "undruggable" targets, for 2 of 6 tested cases. Cryo-EM validation showed sub-angstrom agreement between predicted and experimental structures. Commercially, Chai Discovery secured an Eli Lilly partnership (announced January 2026) described as "one of the pharma industry's largest AI software deals," involving deployment of the Chai platform plus a custom AI model trained on Lilly's proprietary data.
Safety and Optimization
An important caveat: the "before AI" baseline for optimization and safety prediction was never a blank slate. The biopharma industry has used computational and machine learning tools for ADMET assessment for decades. Medicinal chemists applied rule-based filters like Lipinski's Rule of Five and structural alerts for known toxicophores; legacy quantitative structure-activity relationship (QSAR) models from Lhasa Limited (Derek Nexus), Simulations Plus (ADMET Predictor), and others flagged obvious liabilities in silico before compounds were ever synthesized. For lead optimization specifically, tools like Schrodinger's FEP+ (free energy perturbation) have been used since the mid-2010s to predict relative binding affinities, allowing medicinal chemists to computationally triage which analogues to synthesize before committing to wet lab work. Nonetheless, these legacy tools operated within narrow domains, and struggled in tasks requiring multi-objective optimization and in modeling the complex, non-additive interactions between mutations or structural modifications that determine whether a candidate molecule actually works in a living system. Today, frontier AI is able to navigate high-dimensional chemical space in a way these earlier tools could not, learning non-obvious patterns from sparse experimental data that no medicinal chemist could intuit.
Where Chai and Boltz generate candidate molecules from scratch, Cradle occupies the adjacent and arguably more immediately practical problem of lead optimization: taking an existing protein, antibody, or peptide and engineering it to be better across multiple properties simultaneously. The Amsterdam-based company has built a platform that trains on a customer's own experimental data to generate protein variants optimized for specified objectives, then improving with each iterative round of wet lab results; their models can autonomously decide which regions of sequence space to explore, how to balance exploration versus exploitation across candidates, and which mutations to propose, all without the scientist specifying heuristics. In one case study, a top-20 pharma had three late-stage peptide programs that required simultaneous optimization of potency, specificity, expression, and thermostability within tight specification windows, and manual design exhausted all obvious sequence variants that could meet the necessary specifications. In a traditional sequential workflow, a chemist or engineer designs a library of potentially hundreds of variants, sends them to the lab, waits two to four weeks for assay data, discovers that most candidates fail on at least one property, tries to learn from the pattern of failures, designs a new library, and repeats for at least five rounds, requiring months of iteration. Cradle consumed all available sequence-function data from the partner's prior failed rounds, jointly modeled all four properties, and in just a single round, generated at least 48 variants per program that met all four constraints simultaneously. The scientist defined the constraints, selected which prior data to feed the model, evaluated the outputs against their knowledge of the target biology, and decided which candidates to advance, while Cradle took over the combinatorial search that human intuition and iteration simply cannot perform at this dimensionality.
Originally built around biosimulation and regulatory science, Certara has assembled one of the deepest platforms in the industry for pharmacokinetic/pharmacodynamic modeling, physiologically-based pharmacokinetic (PBPK) simulation, and drug-drug interaction prediction. The company's flagship product, Simcyp, has been developed over 25 years through a consortium of 37 pharmaceutical companies and is now recognized and licensed by 11 regulatory agencies worldwide; in August 2025, the EMA formally qualified Simcyp as a trusted engine for DDI projections. The company generated $418.8 million in revenue in 2025 (9% growth), and in October 2025 launched Certara IQ, an AI-powered quantitative systems pharmacology platform designed to make mechanistic modeling accessible beyond the small community of trained pharmacometricians. Certara also recently acquired ChemAxon to expand its computational chemistry capabilities and released Libra, an ML-powered drug-induced liver toxicity Bayesian prediction tool. In March 2026, the FDA accepted Certara's PBPK modeling results in lieu of ten dedicated human clinical pharmacology studies to support the NDA for asciminib (Scemblix), Novartis's chronic myeloid leukemia therapy.
Inductive Bio, a New York-based AI drug discovery company, represents the new generation of ADMET prediction built on large-scale consortium data and modern deep learning rather than expert rules or mechanistic simulation. Inductive's Beacon-1 models won both of the two largest blinded ADMET prediction competitions held to date: the Polaris Ligand ADMET challenge in 2025, and the OpenADMET-ExpansionRx challenge in February 2026. The Inductive approach is built on a pre-competitive data consortium, a growing pool of ADMET data contributed by member pharma and biotech companies, which gives Beacon access to one of the industry's largest and most diverse training sets. The models are then fine-tuned to each partner's specific chemical space through proprietary transfer learning methods. Predictions come with probability estimates rather than binary pass/fail, enabling teams to assess risk on a spectrum, with millions of in silico experiments surfacing the strongest hypotheses for wet-lab validation. In January 2026, Inductive was awarded $21 million from ARPA-H (the Advanced Research Projects Agency for Health) to lead a multi-institutional team, alongside Amgen and several academic partners, in developing next-generation drug toxicity models that improve safety assessment and reduce reliance on animal testing. Unlike first-wave models, Inductive's Beacon models are differentiated in that they learn directly from experimental data at scale, with a continuous feedback loop from partner experiments that updates the models in real time.
Unlike Inductive, which is applying a horizontal approach, aggregating and analyzing data from partners across the ADMET suite, Axiom Bio is going deep on a single problem: drug-induced liver injury (DILI). DILI is responsible for around 20% of drug withdrawals and discontinuations, and has sunk everything from big pharma programs like Pfizer's danuglipron to clinical holds on gene therapies. Axiom Bio co-founders Brandon White and Alex Beatson focused on DILI, raised $15 million in seed funding, and began running their own high-throughput assays on primary human hepatocytes, eventually amassing what Axiom calls the world's largest human toxicity dataset. It contains over 130,000 unique small molecules covering 1,200 targets and 50,000 scaffolds, including specialized classes of compounds like macrocycles, PROTACs, and molecular glues. Each well in Axiom's assays measures 10 to 20 different cellular phenotypes related to toxicity, including apoptosis, necrosis, mitochondrial fission, ER stress, stress granule formation, microtubule stability and then uses imaging and computer vision to process them. Altogether, the company has labeled more than 394 million individual cells and 9 billion mitochondria, which are then paired with a clinical dataset of over 5,200 documented DILI likelihood scores and more than 38,000 liver enzyme elevation data points, allowing Axiom to connect in vitro signals to clinical outcomes at a level of accuracy exceeding the performance of major pharma companies. The company is currently running pilot studies with six of the top twenty pharma companies. A useful comparison here is to Cadence Design Systems, which investor Amplify Partners made at the time of Axiom's seed round: Just as Cadence offers the simulation layer that all semiconductor companies use to validate their designs before sending them off to be fabricated, Axiom will offer the simulation layer that all pharma companies use to validate the safety of a molecule before sending it into an animal and then a clinical trial.
It's worth noting one larger trend: the blurring of the line between "safety prediction" and "molecule design," two different parts of the discovery stack. As generative chemistry platforms begin to incorporate ADMET considerations into their generation loops, the classic stepwise workflow of designing a molecule and then checking if it's safe is being rolled into a single step. For example, Insilico's Chemistry42 platform, a generative chemistry engine, incorporates ADMET filtering into its generative pipeline, which means that the molecules that come out of the platform have already been filtered for predicted liabilities like toxicity, metabolic stability, and solubility before they ever reach the chemist. The question, then, for pure-play ADMET startups is whether they maintain value as a standalone step or are absorbed into the platforms that actually design the molecules themselves.
Yet another often overlooked caveat to the safety and optimization layer hype is that while the models are trained on cellular phenotypes to predict systemic toxicity, the ultimate leap from per-cell signal to whole-organism outcome is significant; for example, liver toxicity in vivo involves not just hepatocyte death but immune response, regeneration capacity, drug-drug interactions, and patient-specific variability in metabolism. A system that performs well at the level of isolated cellular readouts may therefore still miss causal determinants of toxicity in patients.
And amid the proliferation of computational approaches to drug discovery, a contrarian thesis is gaining traction: the rate-limiting step is not prediction but data generation, and the companies most likely to win are those that generate human-relevant data faster, not those that build better models on existing data. As many working at the intersection of AI and biology have noted, while LLMs can be trained on trillions of tokens of internet text generated at near-zero marginal cost, one biological data point can cost thousands of dollars and weeks of labor to produce. Roughly 15% of AI-generated molecules require synthetic routes of more than eight steps (although newer models have begun taking into account real-world manufacturing complexities), and only 60% of disease targets have adequate crystallographic data for structure-based AI design. This isn't a critique of AlphaFold's achievement, which was a revolutionary triumph for scientific and technological progress worldwide, but a calibration of its scope; it showed how far models can go when trained on rich experimental ground truth, but that computation can't necessarily replace the need to generate more of it. Similarly, ADMET models learn from historical assay data; clinical outcome predictors learn from past trials and struggle to anticipate unprecedented mechanisms of action. The only way to expand the frontier is to generate new data, ideally data that is as close to human biology as possible. If this thesis is correct, the most valuable AI applications in biopharma may not be the models themselves but in experimental systems that can accelerate proprietary data generation, creating moats that pure-software players cannot replicate.
2024-2026 marked an inflection point for the role of AI in drug discovery, moving the technology from analytical tool to generative engine. Three developments characterize this shift: First, the architectural stacking of diffusion models, transformers, and graph neural networks into sequential foundation models has enabled researchers to achieve state-of-the-art performance on proteins, small molecules, DNA, RNA, and their interactions all within a single model, and because these models (like Boltz-2) are open source, that capability is available to any scientist in the world. Second, the performance of these models is improving exponentially: Chai-2's 16% hit rate for antibodies; BoltzGen's two-thirds hit rate on novel targets; AlphaProteo's 88% hit rate for binders against viral proteins; Insilico's rentosertib reaching Phase 2a. Third, the rise of end-to-end systems, like IsoDDE's drug design engine or Robin's multi-agent scientific workflow, that turn formerly serial, human steps of drug discovery into continuous computational loops. But importantly, questions and doubts about the future of AI-led discovery remain. No AI-discovered drug has received FDA approval. The hype-to-outcome ratio in AI-native biopharma remains high, and the industry's track record of press-release-driven valuations followed by quiet disappointments should temper enthusiasm. There also remains an unresolved tension re: open-source democratization (Boltz, RFdiffusion, BindCraft) versus proprietary value capture (Isomorphic, Chai-2, Recursion). The operational gains of AI thus far are enormous, yet only time will answer the ultimate question of whether AI can produce better drugs, not just faster ones.